Diciasettesimo Congresso degli Antropologi Italiani
XVII CONGRESSO DEGLI ANTROPOLOGI ITALIANI
ABSTRACTS
- Invited Speakers
- Antropologia Molecolare
- Antropometria
- Biodemografia
- Paleoantropologia e Conservazione
- Sessione sulla Sardegna
- Miscellanea
ANTROPOLOGIA MOLECOLARE
Adattamento genetico-molecolare differenziale a patogeni ambientali: le comunità di pluviselva dell'Ecuador Nord-Occidentale
G.F. De Stefano; F. De Angelis; A. Garzoli
Dipartimento di Biologia, Università di Roma Tor Vergata
Questo studio si riferisce all’analisi molecolare di due comunità residenti nella Provincia di Esmeraldas, nell’Ecuador Nord-Occidentale: l’una di origine africana, l’altra costituita da Amerindi (Indios Cayapa o Chachi). Esse sono risultate geneticamente isolate tra loro nonostante convivano nello stesso ambiente e siano quindi sottoposte ai medesimi agenti selettivi. Un interesse particolare rivestito da queste comunità è quello di rispondere in modo differenziale a vari agenti patogeni, tra cui un nematode parassita: Onchocerca volvulus.
A questo proposito dati di ordine epidemiologica hanno evidenziato una diversa manifestazione clinica della patologia nelle due comunità. Fonti bibliografiche indicano l’influenza degli alleli HLA di classe II nelle infezioni di nematodi parassiti, in particolare è nota una correlazione tra alcune varianti alleliche di tali geni e modificazioni cliniche della patologia provocata da O. volvulus. La presente ricerca è quindi tesa ad evidenziare la presenza di alleli specifici nei soggetti in esame, costituendo il primo screening fra i soggetti sani, individui manifestanti la patologia e soggetti ipoteticamente immuni in entrambe le comunità. A tal fine, l’analisi della distribuzione del linkeage disequilibrium (LD) lungo la regione HLA ha suggerito che alcuni SNPs (single nucleotide polymorphisms) potrebbero agevolare l’analisi della variazione dell’HLA. Alti livelli di LD fra SNPs ed alleli HLA suggeriscono che gli SNPs possano essere informativi riguardo il tipo HLA, quindi SNPs accuratamente scelti possono assicurare la comprensione della variazione in diversi loci. La strategia è perseguita per identificare varianti alleliche dei loci HLA DQA1 e DQB1 nelle due comunità. Fase successiva della ricerca è la tipizzazione molecolare HLA diretta, mediante tecniche SSO (Sequence-Specific Oligonucleotide) e SSP (Sequence-Specific Primers) degli individui che condividono uno stesso pattern polimorfico.
L’analisi degli SNPs, verificati tramite sequenziamento diretto del DNA, non fornisce, in entrambe le comunità, un livello di discriminazione sufficiente tra individui malati e non malati. Viceversa, tramite la tipizzazione diretta degli alleli HLA si evince una discreta eterogeneità allelica tra le due comunità, probabile testimonianza della diversa origine e storia evolutiva e possibile responsabile della risposta differenziale. Nello specifico, all’interno della comunità indiana americana l’allele DQA1*0401 sembra essere presente maggiormente in individui ipoteticamente immuni; mentre nella comunità di origine africana, oltre il DQA1*0401, gli alleli maggiormente rappresentati in individui ipoteticamente immuni risultano essere il DQB1*0301 ed il DQA1*0505 o il DQA1*0509, non univocamente determinabili mediante la metodica utilizzata. Nessun allele risulta, invece, strettamente correlato alla patologia in entrambe le comunità. Tali risultati, seppur parziali, indicano che le varianti alleliche dei geni HLA DQA influenzano il corso dell’infezione da O. volvulus, ed aiutano nel definire uno stato che potrebbe riflettere una immunità genetica nelle comunità in esame.Admixture proportions in southern Malagasy ethnic groups by high-resolution genotyping of maternal and paternal lineages
Stefania Bertoncini (1), Sergio Tofanelli (1), Loredana Castrì (2), Donata Luiselli (2), Francesc Calafell (3), Giuseppe Donati (1), Luca Taglioli (1), Giorgio Paoli (1).
(1) Dipartimento di Biologia, Università di Pisa, Italy. (2) Dipartimento di Biologia Evoluzionistica Sperimentale, Università di Bologna, Italy (3) Department de Ciencies Experimentals de la Salut (CEXS), Unitat de Biologia Evolutiva, Universitat Pompeu Fabra, Barcelona, Spain.
The present population of Madagascar is subdivided into 18 well-defined ethnic groups showing, for morphological and cultural features, different degrees of admixture with Bantu- and Austronesian-speaking populations. A recent genetic study of 4 inland groups (Hurles et al., 2005) demonstrated that African and Indonesian contributions were approximately equal at Y-binary markers and that the Indonesian one is predominant (62%) at mtDNA data.
To more thoroughly investigate the admixture proportions and genetic history of Malagasy people we analysed paternally and maternally inherited lineages of three populations from South-Eastern coasts (Antandroy, Antanosy, Antesaka) and one inland group (Merina) at a more detailed level of phylogenetic resolution. A set of 17 Y-STR loci and 20 Y-binary markers were typed in 110 unrelated males; the HVSI and 17 SNPs from the mtDNA coding region were genotyped in the total sample (N=133).
The proportion of African and Indonesian components from mitochondrial markers was barely the same as in Hurles et al.’s populations (~60% Indonesia and ~40% Africa). However, the hypothesis that migrations from Africa may have been more limited than those from Indonesia does not hold true for coastal groups, as gene diversity was much higher for African-derived maternal lineages.
Y chromosome data gave significantly different component proportions for coastal and inland groups. The majority of coastal paternal lineages were African-derived (Antandroy 86%, Antanosy 64%, Antesaka 62%) and the influence of recent colonizations was not negligible (around 9% of European lineages in Antanosy, 3%).
As a whole, the results point to a common Indonesian genetic matrix as a consequence of a single migration event and to a sex-biased contribution from Africa.
Hurles ME, Sykes BC, Jobling MA, Forster P 2005. The dual origin of the Malagasy in Island Southeast Asia and East Africa: evidence from maternal and paternal lineages. Am J Hum Genet 76:894-901.Analisi della struttura genetica della popolazione corsa e sarda
Laurent Varesi (1), Alessandra Falchi (1), Pedro Moral (2), Maria E Ghiani (3), Ignazio Piras (3), Carla M Calò (3), Giuseppe Vona (3)
(1) Laboratoire de génétique humaine, Faculté des sciences, Université de Corse (F) (2) Departament de biologia animal-antropologia. Facultat de Biologia, Barcelona (E) (3) Dipartimento di Biologia sperimentale, Università di Cagliari
In questo studio sono stati analizzati 18 marcatori eritrocitari e plasmatici a partire da diverse centinaia di individui provenienti da differenti micro-regioni della Sardegna (Gallura, Nuorese, campidano di Cagliari) e della Corsica (Nord est, centro, Nord ovest, Sud ovest e estremo Sud).
Le analisi mostrano una debole influenza delle popolazioni continentali sulla struttura genetica delle due popolazioni insulari e una similitudine genetica delle popolazioni corse e sarde. Queste similitudini si rispecchiano nella distribuzione delle mutazioni responsabili delle beta-talassemie.
La stessa mutazione (beta-39) è in prevalenza associata a questa patologia nelle due isole. L’analisi degli aplotipi della zona della famiglia delle beta-globine, ha rivelato una origine comune di questa mutazione, che riflette una possibile origine comune.
L’analisi del DNA mitocondriale mostra le stesse similitudini genetiche. Questo studio ha inoltre apportato qualche precisione sulla storia demografica delle popolazioni insulari e sul tempo di espansione del loro popolamento.
Questa ricerca è stata realizzata grazie ai fondi della comunità europea (Programmes INTERREG).Analisi della variabilità del locus - 13910 associato alla tolleranza al lattosio in Europa meridionale
Paolo Anagnostou (1), Cristina Fabbri (2), Cinzia Battaggia (1), Valentina Coia (1), Davide Pettener (2), Cristian Capelli (3), Margarida Coelho (4), Jorge Rocha (4), Giovanni Destro-Bisol1 (5) e Donata Luiselli (2)
(1) Dipartimento di Biologia Animale e dell’Uomo, Università di Roma“La Sapienza” (2) Dipartimento di Biologia Evoluzionistica Sperimentale, Università di Bologna (3) Dipartimento di Zoologia, Università di Oxford, UK (4) Istituto di Patologia e Immunologia Molecolare (IPATIMUP), Università di Porto, Portogallo (5) Istituto Italiano di Antropologia, Roma
La capacità di digerire il lattosio in età adulta è una condizione ereditaria causata dalla persistenza dell’enzima Lattasi Florizin Idrolasi dopo lo svezzamento. Numerosi studi hanno dimostrato come la distribuzione della tolleranza al lattosio sia estremamente varia tra le popolazioni umane. Tale variabilità è stata oggetto di diverse ipotesi sulla sua evoluzione e sul ruolo che la selezione naturale ha avuto nel generare la situazione osservata attualmente. L’identificazione della mutazione T-13910 associata alla tolleranza al lattosio, nelle popolazioni nord-europee, ha aperto nuove frontiere nello studio dell’evoluzione di tale condizione fisiologica.
Nel presente lavoro è stata studiata la variabilità microsatellite associata alla mutazione T-13910 in Italia e Grecia allo scopo di inferire due parametri chiave dell’evoluzione della tolleranza al lattosio: l’azione della selezione naturale e l’età della mutazione.
Sono stati utilizzati un totale di 749 campioni provenienti dall’Italia e 100 campioni di provenienza greca. All’estrazione del DNA ha fatto seguito l’amplificazione mediante PCR e il taglio enzimatico per la discriminazione degli alleli C e T al locus C/T-13910. Gli alleli STR ai loci D2S3013, D2S3015 e D2S3010 sono stati tipizzati tramite sequenziatore semiautomatico (ALF). Le fasi gametiche sono state inferite con il software Phase 2.1. L’età del MRCA è stata calcolata utilizzando il metodo proposto da Coelho et al (2005), per valutare l’azione della selezione naturale è stato utilizzato il metodo proposto da Slatkin e Bertorelle (2001).
I dati relativi della tolleranza al lattosio predetta dalla mutazione T-13910, confermerebbero il dato osservato, tramite test fisiologici, dell’andamento clinale della tolleranza al lattosio in Europa. L’associazione di una ristretto numero di alleli STR, simili in tutte le popolazioni esaminate, con la mutazione T-13910, e la presenza del medesimo aplotipo composito modale in tutte le popolazioni, suggeriscono un’origine unica di tale mutazione.
La bassa variabilità osservata nei loci STR associati alla veriante T-13910 suggerisce un’origine recente di tale mutazione. Il test di neutralità selettiva mostra la presenza di effetti selettivi principalmente in Italia.
Presi nel loro insieme, questi risultati suggeriscono che la tolleranza al lattosio, associata alla mutazione T-13910, sia un tratto sorto in tempi relativamente recenti (circa 30.000 – 10.000 anni fa) e avvalorano l’ipotesi che l’attuale distribuzione di questo genotipo risulti, almeno in parte, dal vantaggio selettivo da esso conferito in termini di migliore utilizzazione delle proprietà nutrizionali del latte.Correlation of genetic, geographic and linguistic distances in Bosnia and Herzegovina
Naris Pojskic (1), Lejla Kapur (1), Damir Marjanovic (1), Hadziselimovic R. (1,2)
(1) Institute for Genetic Engineering and Biotechnology, University of Sarajevo (2) Biology Department, Faculty of Science, University of Sarajevo
Anthropogenetic researches in Bosnia and Herzegovina started in middle of 19th century by Austro-Hungarian physicians who have done their researches during recruitment of Bosnian young people for Imperial army services. Main goal of these researches were to estimate differences between three ethnic (in that time described as only religious) groups in Bosnia and Herzegovina: Orthodox Christians, Catholic Christians and Muslims. Colors of eyes and hair together with various morphological traits have been used as the marker. During the 20th century the most researches were done on local human populations, which have geographic specificity with expressed of patrilocality. Several biochemical-physiological (ABO system, PTC tasting sensitivity, secretor/nonsecretor, green ad red color visions etc.) and various morphological traits have been observed.
At the end of 20th and beginning of 21st century during researches of human populations in Bosnia and Herzegovina DNA markers have been introduced (autosomal STR, mtDNA HVS-I and HVS-II, Y-chromosomal markers). Beside observations of isolated local human population, the genetic structure of three main ethnic (national) groups in Bosnia and Herzegovina have been investigated. According the B&H Constitution in Bosnia and Herzegovina exist three main ethnic (national) community with the same rights and obligations. These groups speak three languages: Bosnian, Croatian and Serbian.
We analyzed possible correlation between Reynold’s genetic distance, geographical and linguistic distance among three main ethnic (national) community observing expressed patrilocality populations spread in 10 different regions. Since three B&H languages belong to the same language group, linguistic “micro”-distance was estimated as proportion of different words (some languages have more influences by foreign languages during B&H history.
Ordinary approaches for geographic distance estimation were not applied. It has been proved that “shortest road distance” is more appropriate for estimating geographical distance for this research, since the having the road from the isolated places mean possible migration. For estimation of correlations between distances Mantel test and coefficient of distance have been implemented. Estimation were done based on data obtained from applications of various markers. The results imply that there is no significant correlation between genetic and linguistic distance among three main ethnic (national) B&H groups. Although, it is noticed that generally small figures of genetic distance (based on variation of STR and biochemical-physiological and morphological traits) among populations and groups estimation “follow” small linguistic difference.
The both implemented measures show no significant correlation between genetic and geographic distance among populations in different area in Bosnia and Herzegovina.Dispersal patterns of M267-derived Y chromosomes in the Mediterranean
Sergio Tofanelli (1), Gianmarco Ferri (2), Laura Caciagli (1), Luca Taglioli (1), Donata Luiselli (3), Giorgio Paoli (1), Cristian Capelli (4)
(1) Dipartimento di Biologia, Università di Pisa, Italy. (2) Laboratorio di Medicina Legale, Università di Modena e Reggio Emilia, Italy. (3) Dipartimento di Biologia Evoluzionistica Sperimentale, Università di Bologna, Italy (4) Department of Zoology, University of Oxford, UK
Human Y chromosomes belonging to haplogroup J1 (International Society of Genetic Genealogy 2007) share a derived state (G) at the M267 mutation site. It has been argued (Semino et al., 2004) that this mutation originated some 24,000 years ago in the Near East or North-East Africa and spread in the Mediterranean by means of at least two temporally distinct migrations: the first would have occurred towards Aegean and Italian coasts in Neolithic times; a more recent one (estimated time bounds 8.7–4.3 Ky) would have diffuse M267-G in Northern Africa. According to other authors (Nebel et al., 2001; Al-Zahery et al., 2003; Di Giacomo et al., 2004), however, M267-G would have arisen as early as 10,000 years ago and would mark the historical expansion of Arabian tribes in the northern Levant and southern Africa.
We investigated the variability of M267-G chromosomes from 23 different Mediterranean populations (original and published data) at a total of 20 Y STR loci. Three different sets of markers were considered: the “Y-filer” set (DYS456, DYS389I, DYS390, DYS389II, DYS458, DYS19, DYS385a, DYS385b, DYS393, DYS391, DYS439, DYS635, YGATA-H4, DYS437, DYS392, DYS438, DYS448) allowed to more accurately reconstruct time and space of the main dispersal events associated with this mutation; the “MH” (DYS19, DYS388, DYS390, DYS391, DYS392, DYS393) and the “DL3” (DYS388, YCAIIa, YCAIIb) sets allowed the origin and diffusion of local modal haplotypes to be better defined.
The results depict a more complex and deeper stratification of haplotype-clades than thought before. In fact, we could observe both, geographically structured and even lineages, that could be associated to pre-agricultural, Neolithic and historical demographic events.
Al-Zahery N, Semino O, Benuzzi G et al. 2003. Y-chromosome and mtDNA polymorphisms in Iraq, a crossroad of the early human dispersal and of post-Neolithic migrations. Mol Phylogenet Evol 28:458-472.
Di Giacomo F, Luca F, Popa LO et al. 2004 Y chromosomal haplogroup J as a signature of the post-neolithic colonization of Europe. Hum Genet 115:357–371.
International Society Of Genetic Genealogy 2007. Y-DNA Haplogroup Tree 2006. Version: 1.24, Date: 7 June 2007, http://www.isogg.org/tree/Main06.html.
Nebel A, Landau-Tasseron E et al. 2002. Genetic evidence for the expansion of Arabian tribes into the Southern Levant and North Africa. Am J Hum Genet 70:1594–1596.
Semino O, Magri C, Benuzzi G et al. 2004. Origin, diffusion, and differentiation of Y-chromosome haplogroups E and J: inferences on the Neolithization of Europe and later migratory events in the Mediterranean area. Am J Hum Genet 74:1023–1034.Filogeografia dell'aplogruppo B del cromosoma Y in Africa Centrale attraverso uno studio ad alta risoluzione della variabilità microsatellite
Chiara Batini (1,2), Gemma Berniell-Lee (2), Francesco Donati (1), Gabriella Spedini (1,3), Cristian Capelli (4), David Comas (2), Giovanni Destro-Bisol (1,3)
(1) Dipartimento di Biologia Animale e dell’Uomo, Sapienza Università di Roma, Roma, Italia (2) Unitat de Biologia Evolutiva, Universitat “Pompeu Fabra”, Barcelona, Spagna (3) Istituto Italiano di Antropologia, Roma, Italia (4) Department of Zoology, University of Oxford, Oxford, UK
L'aplogruppo B rappresenta uno dei rami più antichi dell'albero filogenetico del cromosoma Y. La sua distribuzione, limitata all'Africa sub-sahariana, è particolarmente interessante per la presenza, non solo in popolazioni di agricoltori e pastori, ma anche in tutte le popolazioni di cacciatori-raccoglitori analizzate finora: i Kung del Sud Africa, i Pigmei Mbuti della Repubblica Democratica del Congo ed i Pigmei Biaka della Repubblica Centrafricana. Questo aplogruppo sembra quindi essere un carattere plesiomorfo delle popolazioni dell'Africa sub-sahariana.
Lo scopo del presente lavoro è studiare in dettaglio la distribuzione dell'aplogruppo B in popolazioni dell'Africa Centrale, attraverso l'analisi di 7 distinte popolazioni di Pigmei Occidentali (2 Baka e 2 Bakola del Camerun, Babinga della Repubblica Popolare del Congo, Baka del Gabon e Mbenzele della Repubblica Centrafricana), e di 20 gruppi di lingua Bantu. L'analisi di tale aplogruppo in queste popolazioni offre la possibilità di raccogliere informazioni importanti per la ricostruzione del popolamento antico di questa area, tuttora poco studiato a causa dell'assenza di resti fossili e della difficile datazione dei resti archeologici.
Lo screening di queste popolazioni per il polimorfismo M60 (che definisce l'aplogruppo B) ha portato alla selezione di 153 individui appartenenti a questo aplogruppo, successivamente tipizzati per 12 polimorfismi STR (DYS19, DYS385a, DYS385b, DYS389I, DYS389II, DYS390, DYS391, DYS392, DYS393, DYS437, DYS438, DYS439). I dati raccolti sono stati analizzati attraverso un median-joining network utilizzando gli aplotipi STR ed applicando un approccio filogeografico. Abbiamo infine confrontato i dati ottenuti per il cromosoma Y con quelli dell'aplogruppo L1c del DNA mitocondriale, mettendo in evidenza alcune analogie nella distribuzione dei due aplogruppi. In tal modo abbiamo potuto analizzare le relazioni evolutive esistenti tra i vari gruppi pigmei e le popolazioni di lingua Bantu considerando lineages a trasmissione sia maschile che femminile.Identificazione di alleli rari nel Nord Sardegna attraverso l’analisi dell’AmpFℓSTR® Yfiler™ PCR Amplification Kit
Alessandro Mameli (1), Giuseppe Vona (2), Gavino Piras (1), Andrea Berti (3), Maria Elena Ghiani (2)
(1)Reparto Carabinieri Investigazioni Scientifiche (Cagliari) (2)Dipartimento di Biologia Sperimentale. Università di Cagliari (3)Reparto Carabinieri Investigazioni Scientifiche (Roma)
L’utilizzo dei marcatori del cromosoma Y per studi di genetica di popolazione umana e analisi forensi è ormai di uso frequente. Gli aplotipi costruiti con i marcatori dell’Y possono essere usati per studi di linee paterne, per differenziare gruppi di popolazioni umane, e per analisi forensi.
In questo lavoro abbiamo analizzato un totale di 100 soggetti maschi, sani, autoctoni del Nord-Sardegna, non imparentati fra loro attraverso il kit di amplificazione AmpFISTRs Yfiler.
Questo sistema amplifica simultaneamente 17 loci Y-STR, che includono l’European minimal haplotype, e i loci STR raccomandati dallo Scientific Working Group on DNA Analysis Methods (SWGDAM), oltre ai loci DYS437, DYS448, DYS456, DYS458, Y GATA H4, e DYS635.
Il DNA è stato estratto da sangue seguendo il metodo di Gill et al. (1985). L’amplificazione del DNA è stata eseguita con il AmpFlSTR Yfiler PCR amplification kit in un Geneamp®PCR System 9700, usando i criteri suggeriti dalla casa produttrice. La separazione e identificazione del prodotto di PCR degli Y-STR 17plex è stata effettuata tramite ABI Prism 3100 Avant Genetic Analyzer. Per l’attribuzione degli alleli specifici per ciascun locus sono stati usati dei ladder allelici forniti con il kit (AmpFlSTRYfiler allelic ladder).
L’analisi dei singoli campioni analizzati in questo lavoro ha messo in evidenze alcune particolarità della popolazione del Nord Sardegna. Sono stati osservati alcuni alleli che non sono inclusi nel ladder allelico fornito nel kit di amplificazione dell’AmpFℓSTR® Yfiler™ per esempio l’allele DYS458*11 che si presenta con una frequenza di 0,01. Il locus DYS458 mostra un allele (DYS458*17.2) in una forma non-consensus con una frequenza anche esso di 0,01. Un altro allele non-consensus (11.2) si è presentato all’analisi del locus DYS438. Un lavoro su una popolazione del Portogallo (Alves et al., 2007) ha mostrato una interessante associazione tra il locus DYS458 e il paragruppo J*(xJ1,2), infatti tutti gli individui caratterizzati da alleli non-consensus per il DYS458 all’analisi con polimorfismi Y-SNP sono risultati appartenenti al paragruppo J*(xJ1,2). Questo potrebbe indicare che il DYS458 potrebbe essere usato per discriminare un aplogruppo all’interno di un paragruppo.
La maggior parte degli Y-STR utilizzati per l’analisi forense (DYS19, DYS389 I and II, DYS390, DYS391, DYS392, DYS393 and DYS385) hanno riportato doppi alleli per un singolo locus a causa di eventi di duplicazioni seguiti da mutazione. La presenza di doppi alleli potrebbe significare la presenza di più profili maschili in uno stesso campione, ed è per questo motivo che è importante conoscere l’incidenza di duplicazioni-mutazioni per un locus all’interno di una popolazione. Nel nostro campione del Nord Sardegna abbiamo trovato una duplicazione al locus DYS19 con gli alleli 15/16 su un individuo. L’individuazione di particolari forme alleliche è di grande interesse sia da un punto di vista di genetica di popolazione, perché tipica di determinate popolazioni sia per le indagine forensi perché di grande aiuto nella caratterizzazione dei campioni esaminati.Il Dawro Konta: primi dati sulle popolazioni etiopi della valle dell’Omo
Manuela Cioffi, Loredana Castrì, Davide Pettener, Donata Luiselli
Laboratorio di Antropologia molecolare, Dipartimento di Biologia evoluzionistica sperimentale, Università di Bologna
Questo studio si propone di esaminare la variabilità del patrimonio genetico di un piccolo gruppo della popolazione Dawro nella valle dell’Omo in Etiopia. L’etnia Dawro fa parte del ceppo omotico, appartenente alla famiglia linguistica Afro-Asiatica. Diversamente da Amhara ed Oromo, popolazioni di questo ceppo linguistico non sono mai state studiate dal punto di vista genetico-molecolare; inoltre per secoli questa zona è rimasta pressoché inaccessibile.
La lingua omotica è considerata la ramificazione più divergente dell’Afro-Asiatico; scopo di questa ricerca è verificare inoltre se la divergenza linguistica trova riscontro nelle caratteristiche genetiche della popolazione Dawro.
Il campionamento è stato eseguito presso la missione dei Frati Cappuccini dell’Emilia-Romagna del villaggio di Gassa Chare (2500 m s.l.m.), regione Dawro, Etiopia. Mediante un prelievo di cellule di mucosa buccale sono stati analizzati 39 soggetti sani, di età, sesso e clan assortiti, tutti di etnia Dawro, appartenenti allo stesso villaggio. Tutti i soggetti, mediante la compilazione di una scheda che raccoglie dati biodemografici, hanno dato il consenso di partecipazione alla ricerca. Le prime analisi hanno riguardato oltre l’estrazione del DNA mediante protocolli standard, l’amplificazione della regione HVSI del genoma mitocondriale (15996-16401). Il sequenziamento è stato effettuato mediante sequenziatore ABI PRISM 3730 (Applied Biosystem).
Inoltre sono stati testati tre siti di restrizione (+ 3592HpaI, + 10871MnlI, +10397AluI) per identificare rispettivamente i clusters L1/L2, N ed M.
Le analisi di restrizione condotte su tutti i 39 soggetti campionati hanno evidenziato le seguenti frequenze per cluster: L1/L2 (28%), N (10%), L3 (49%), M (13%). I risultati preliminari riguardo la sequenza della regione di controllo evidenziano la presenza dei seguenti aplogruppi: L0 (subclade L0a2), L2 (L2a1c), L3 (L3x, L3x2), L5 (L5a1, L5a2) e K.
Le regioni dell'Africa orientale sono caratterizzate, dal punto di vista genetico, in modo ancora incompleto. La ricostruzione della storia delle popolazioni etiopi è complicata dal fatto che probabilmente hanno ricevuto in passato un flusso genico proveniente da altre popolazioni già ibride.
La metà degli Etiopi finora studiati ha linee mitocondriali che appartengono a cladi specifici dell'Africa sub-sahariana, mentre l'altra metà si divide tra subcladi derivati dagli aplogruppi M e N che sono molto comuni fuori dall'Africa. In particolare, da studi effettuati da Kivisild et al., (2004) in Etiopia, risulta che le frequenze relative ai raggruppamenti L1/L2, N, L3 e M sono rispettivamente 28,8%, 30,8%, 23,4% e 17%.
Dal confronto del tutto preliminare tra questi dati e i risultati ottenuti dalla presente ricerca, si nota nella popolazione del Dawro Konta un contributo degli aplogruppi N ed M minore rispetto alla media etiopica e una maggiore frequenza del clade L3, che fa pensare ad un minor flusso genico da parte di gruppi di origine euroasiatica.Il Kurdistan iracheno: variabilità di marcatori autosomici (Alu) e uniparentali (cromosoma Y e DNA mitocondriale)
Antonella Useli (1), Loredana Castrì (1), Cristina Fabbri (1), Stefano Benazzi (2), Gianmarco Ferri (3), Davide Pettener (1), Donata Luiselli (1)
(1) Dipartimento di Biologia evoluzionistica sperimentale, Laboratorio di Antropologia molecolare, Università di Bologna (2) Dipartimento di Storie e Metodi per la Conservazione dei Beni Culturali, Università di Bologna (3) Dipartimento integrato di Servizi Diagnostici e di Laboratorio, Università di Modena e Reggio Emilia
Il Kurdistan occupa una vasta area tra la Mesopotamia, l’Anatolia e l’altopiano iranico. Questa regione non ha mai avuto un’unità politica e nel 1923 è stata annessa a quattro stati (Iraq, Iran, Turchia e Siria), pur ospitando una popolazione, numericamente rilevante, caratterizzata da una forte unità culturale e linguistica. L’origine di questa popolazione di lingua indoeuropea non è stata ancora chiarita e la popolazione curda non è mai stata studiata in modo dettagliato dal punto di vista genetico. L’analisi della variabilità di marcatori molecolari a trasmissione uniparentale, come il cromosoma Y (NRY) ed il DNA mitocondriale (mtDNA), e di sistemi autosomici, come i marcatori Alu, può essere considerata uno strumento utile per rilevare i fenomeni microevolutivi che hanno caratterizzato la storia genetica delle popolazioni e che in relazione con i dati storici, linguistici ed etnologici possono aiutare a comprenderne l’origine e la storia.
L’obiettivo di questa ricerca è: 1) ottenere un quadro completo della variabilità genetica presente in un campione di individui curdi; 2) valutare le relazioni filogenetiche con le altre popolazioni mediorientali; 3) chiarire se la forte connotazione etnica dei Curdi si riflette anche nella condivisione di un pool genetico omogeneo.
Il campione di popolazione studiato è costituito da 71 individui: 49 di etnia curda, provenienti prevalentemente dal Kurdistan iracheno, e 22 di etnia persiana, provenienti dall’Iran sud occidentale. L’analisi è stata condotta utilizzando 8 polimorfismi di inserzione Alu autosomici, l’analisi di sequenza dell’HVSI e 13 RFLPs della regione codificante del mtDNA e 44 marcatori (33 SNPs e 11 STRs) sulla regione non ricombinante del cromosoma Y (NRY). Avvalendoci dei dati sul campione persiano da noi analizzato e di quelli presenti in letteratura e utilizzando un approccio di tipo filogenetico e filogeografico, è stato possibile inserire la variabilità osservata nel contesto eurasiatico e mediorientale.
Il campione di individui curdi studiato, considerato in un contesto macrogeografico, si colloca nell’ambito della variabilità eurasiatica occidentale e, in un ambito geografico più ristretto, rientra nei pattern di variabilità dell’Asia occidentale e mediorientale.
Nell’indagine a livello microgeografico il DNA mitocondriale ed il cromosoma Y hanno mostrato un potere informativo più elevato rispetto a quello dei polimorfismi Alu, in particolare quando si utilizzano i dati sulla distribuzione degli aplogruppi.
La presenza, nel campione curdo analizzato, dei principali aplogruppi mitocondriali e del cromosoma Y diffusi in Medio Oriente ed pattern di variabilità osservati, sottolineano l’affinità con le popolazioni geograficamente vicine e suggeriscono la condivisione dei principali eventi di popolamento della regione. Le analisi condotte, ed in particolare il confronto con altri gruppi curdi provenienti da altre regioni geografiche, sottolineano tuttavia la complessità dei processi microevolutivi che hanno coinvolto i Curdi e le altre popolazioni mediorientali.Isolamento genetico e isolamento culturale nei Baschi: analisi di una regione di 1 Mb priva di geni del cromosoma 22
Paolo Garagnani (1), Francesc Calafell (2), Anna Gonzales-Neira (2), Donata Luiselli (1), Jaume Bertranpetit (2)
(1) Lab. di Antropologia molecolare, Dipartimento di Biologia Evoluzionistica Sperimentale (BES), Università di Bologna - (2) Unitat de Biologia Evolutiva Facultat de Ciències de la Salut i de la Vida, Universitat Pompeu Fabra, Barcelona, Catalonia
Attualmente è possibile analizzare la variabilità dell’intero genoma in termini di single nucleotide polymorphisms (SNPs) su singoli arrays, riducendo notevolmente costi e tempi di analisi. Queste nuove applicazioni forniranno un importante contributo nel delineare le basi molecolari delle malattie complesse. Nelle popolazioni geneticamente isolate i segnali di linkage disequilibrium (LD) sono più ampi, e i livelli di eterozigosità sono più contenuti, rendendole particolarmente adatte a questo genere di analisi.
Nel presente studio abbiamo voluto verificare se due popolazioni basche, generalmente considerate isolate nel contesto europeo sulla base di analisi condotte su diversi marcatori genetici, confermavano tale isolamento anche analizzando un’ampia regione (1-Mb) priva di geni sul cromosoma 22. A tale scopo sono stati tipizzati numerosi SNPs (single nucleotide polymorphisms), non solo nelle due popolazioni basche provenienti da Spagna e Francia, ma anche in altri gruppi di popolazione spagnola e tre campioni nord-africani, per verificare il contributo di quest’ultime nel determinare le caratteristiche del pool genico delle attuali popolazioni spagnole.
A tele scopo sono stati tipizzati 541 individui appartenenti alle 8 popolazioni. Sulla base della distanza fisica sono stati selezionati 211 SNPs in modo da ottenere una griglia di marcatori quanto più possibile omogenea. La genotipizzazione è stata realizzata su piattaforma Sequenom. Nell’analisi finale sono stati considerati 123 SNPs con una media di 1 SNP ogni 8030 bp. I dati ottenuti sono stati analizzati mediante i software Arlequin, Statistica, Haploview e Structure.
I valori di eterozigosità osservati hanno mostrato un range di variabilità ristretto tra tutte le popolazioni considerate. Come era da attendersi, le popolazioni africane riportano i valori più elevati, ma se si considerano gli errori standard gli indici di eterozigosità si sovrappongono ampiamente fra tutte le popolazioni. Le due popolazioni basche presentano i valori maggiori all’interno della penisola iberica.
L’analisi AMOVA condotta considerando diversi tipi di raggruppamento ha sempre evidenziato valori di Fst e di Fct bassi e non significativi all’interno dei raggruppamenti continentali. La matrice di distanza rappresentata mediante scaling multidimensionale non metrico evidenzia infatti soltanto differenze tra i due gruppi principali (3 popolazioni africane e 5 iberiche).
L’analisi delle dimensioni dei blocchi di linkage conferma l’omogeneità emersa anche dalle altre analisi, con valori più elevati per la popolazione catalana, mentre le popolazioni basche non presentano i valori attesi di LD più ampi rispetto alle altre popolazioni. Soltanto 14 dei 123 SNPs riportano valori di Fst > 0.05 e con tali marcatori è possibile distinguere una differenziazione tra Iberici e nord Africani, mentre i rimanenti loci non sono in grado di evidenziare una sub struttura genetica.
In conclusione, le popolazioni basche non presentano le caratteristiche tipiche delle popolazioni isolate tali da renderle preferibili per studi di associazione.Kenyan crossroads: migrazione e flusso genico in sei gruppi etnici dell'Africa orientale
Loredana Castrì, Paolo Garagnani, Antonella Useli, Mariangela Laino, Elena Flamigni, Davide Pettener, Donata Luiselli
Dipartimento di Biologia evoluzionistica sperimentale, Laboratorio di Antropologia molecolare, Università di Bologna
Il DNA mitocondriale umano presenta diverse caratteristiche (elevato numero di copie, eredità materna, assenza di ricombinazione e alto tasso di mutazione) che lo rendono un marcatore particolarmente utile ai fini dello studio dell'origine e dei patterns di espansione delle popolazioni umane. La presente ricerca riguarda l’analisi della variabilità mitocondriale in sei gruppi etnici del Kenya. La regione, situata nella parte nord-orientale dell’Africa, ha probabilmente giocato un ruolo molto importante nell’origine dell’uomo moderno. I dati paleoantropologici e archeologici indicano infatti l’Africa orientale come il probabile luogo d’origine dal quale l’uomo moderno avrebbe iniziato il suo viaggio di colonizzazione dei diversi continenti. In tempi più recenti, negli ultimi 5000 anni, il Kenya è stato interessato da diversi movimenti migratori di popolazioni provenienti dalle regioni dell’Africa occidentale e settentrionale. Attualmente il territorio è popolato da più di 30 etnie che parlano lingue appartenenti alle tre famiglie linguistiche Niger-Kordofoniana, Afro-Asiatica e Nilo-Sahariana.
Lo scopo principale di questo lavoro è analizzare, utilizzando un approccio filogeografico, la diversità genetica presente in sei popolazioni del Kenya per verificare la presenza di antiche linee mitocondriale africane e valutare l’eventuale ruolo di questa regione nella etno-genesi delle attuali popolazioni africane.
Il campione esaminato è costituito da 286 donatori maschi non imparentati per via materna. Sono state analizzate le regioni ipervariabili I e II (HVSI/HVSII) del DNA mitocondriale e i siti RFLP sulla regione codificante che permettono di individuare gli aplogruppi mitocondriali africani ed eurasiatici. Gli aplotipi mitocondriali osservati in Kenya sono stati quindi confrontati con quelli presenti in un database di circa 100 popolazioni africane e mediorientali allo scopo di valutare le relazioni filogenetiche dei gruppi etnici del Kenya con le altre popolazioni dell’Africa sub-sahariana e in particolare con quelle dell’Africa orientale e della vicina regione mediorientale.
I risultati ottenuti evidenziano la presenza nei sei gruppi etnici kenioti di aplogruppi mitocondriali di origine sia africana che eurasiatica. È stata rilevata una sostanziale omogeneità genetica delle popolazioni analizzate, con l’eccezione del gruppo etnico di pescatori ElMolo che risultano caratterizzati da un forte effetto fondatore e probabile isolamento genetico. L’omogeneità genetica delle altre popolazioni prese in esame è probabilmente da imputare alla presenza di un continuo flusso genico tra le diverse etnie che ha portato ad una somiglianza maggiore di quella ipotizzabile sulla base degli eventi storici e dei conflitti etnici che hanno interessato e continuano ad interessare questa regione. Inoltre, l’analisi filogeografica ha permesso di evidenziare l’esistenza di flusso genico con altre popolazioni dell’Africa orientale, in particolare Etiopi e Bantu.La domesticazione dei bovini italiani: ipotesi suggerite dallo studio paleogenetico di antichi reperti di Bos primigenius
Giulio Catalano (1), Martina Lari (1), Elena Pilli (1), Lucio Milani (1), Paolo Boscato(2), Carolina Di Patti (3), Luca Sineo (4), Carles Lalueza Fox (5), David Caramelli (1).
(1) Dipartimento di Biologia animale e Genetica “Leo Pardi”, Università di Firenze, Italia (2) Dipartimento di Scienze Ambientali, Università di Siena, Italia (3) Museo di Geologia e Paleontologia "G. G. Gemmellaro", Università di Palermo, Italia (4) Dipartimento di Biologia animale “Giuseppe Reverberi”, Università degli Studi di Palermo, Italia (5) Dipartimento di Biologia evolutiva -UPF – Barcelona, Spagna
Lo sviluppo di sempre più sofisticati protocolli di estrazione, amplificazione e sequenziamento di DNA da reperti molto antichi ha incoraggiato l’intervento della genetica nella questione della domesticazione, di specie animali e vegetali. Negli ultimi anni l’attenzione si è focalizzata, in particolar modo, sull’origine e sull’evoluzione di un processo molto complesso, la domesticazione dei bovini europei. Evidenze archeologiche collocano, intorno ai 10000 anni fa, nell’area identificabile con la Mezza Luna Fertile, il centro di espansione della varietà domestica, oggi diffusa in tutta Europa, Bos taurus . In accordo con il dato archeologico, l’analisi molecolare di specifici marcatori genetici (mtDNA, chrY) ha mostrato una variazione della popolazione bovina europea più bassa di quella dei gruppi del Medio Oriente, supportando così l’ipotesi che essa sia derivata dall’espansione neolitica della pastorizia verso Ovest attraverso l’Europa.
Per comprendere meglio le traiettorie di tale processo è stato di fondamentale importanza chiarire la relazione filogenetica esistente tra la forma selvatica, Bos primigenius, e quella attuale.
L’analisi del DNA antico, estratto da bovini inglesi e continentali, ha permesso di stabilire che gli Uri selvatici non sembrano essere i fondatori dei bovini addomesticati che abbiamo oggi.
Alla luce di queste scoperte, l’obbiettivo del nostro lavoro è stato quello di riscontrare, immaginando la possibilità nel passato di incroci spontanei o ad opera dell’uomo, un contributo ancestrale nel pool mitocondriale delle varietà italiane moderne. I dati forniti dalle nostre indagini sperimentali si sono rivelate inaspettatamente in controtendenza al modello continentale osservato, dove esiste una differenza marcata tra la specie primigenia e le razze attuali. In Italia infatti campioni molto antichi presentano le stesse identiche sequenze che oggi ritroviamo sia tra le razze italiane sia tra quelle europee. Con questi risultati è lecito ipotizzare che forse la domesticazione non sia avvenuta in un solo momento e in un solo luogo come sostenuto sino ad oggi. Oppure come nel caso di altre faune italiane, è possibile che ci sia stato un effetto di isolamento dovuto alla presenza di barriere geografiche (Alpi), che hanno determinato la riduzione della variabilità genetica nei Bos primigenius italiani, come quella prodotta più tardi, in età neolitica, dalla selezione imposta dai primi allevatori.La malattia celiaca. Aspetti genetici e antropologici
Luca Sineo e Matilde Passantino
Dipartimento di Biologia animale “G. Reverberi”- Università degli Studi di Palermo/Via Archirafi 18, 90123 Palermo – llsineo@unipa.it
L’intolleranza al glutine può a buon titolo includersi tra le patologie da transizione ecologica che interessano l’uomo moderno. La base genetica di questo stato morboso è stata da lungo tempo messa in relazione ad alcuni aplotipi del sistema HLA. Questo ha portato in tempi recenti alla presunta identificazione di alcuni aplotipi “predisponenti” in diverse aree geografiche ed in diverse popolazioni: l’aplotipo identificato nei caucasici celiaci del nord europa è A1 B8 DR3 DQ2, quello del sud europa A1B8DR3 DQ2/DR7 DQ2 o DR5 DQ7/DR7 DQ2; abbiamo un aplotipo “sardo” A30 B18 DR3 DQ2, Punjabi A26 B8 DR3 DQ2 e Ax B21 DR3 DQ2, e “anatolico” A2 B8 DR3 DQ2.
Questa oggi suona come una banalizzazione, data la palese eziologia multifattoriale e l’esistenza di diversi geni candidati che concorrono a questo stato morboso (CD14, CTLA4, MIC, IL2, IL21).
Comunque, il ruolo degli aplotipi di seconda classe DRx -DQ2 e/o DQ8 nella presentazione dell’antigene Gliadina + Transglutaminasi, quantomeno quindi in una fase dell’intolleranza e della sua evoluzione clinica, è indubbio.
Sono stati esaminati dati relativi a 225 gruppi etnici o popolazioni geograficamente localizzabili (2000 voci bibliografiche dal 1973 al 2006 - nonché il DBase http://www.allelefrequencies.net). Di 120 di queste popolazioni, comparabili per tipizzazione, è stato preparato un report correlato da mappe di distribuzione (Microsoft Mappoint 2006). Per motivi di standardizzazione e di analisi i dati sono stati preventivamente suddivisi in tre gruppi a seconda della tipizzazione.
I gruppo
DRB1*03-DQA1*05 – DQB1*02
DRB1*04-DQA1*03 – DQB1*0302
DRB1*07-DQA1*0201 – DQB1*02
II gruppo
DQA1*05-DQB1*02
DQA1*03-DQB1*0302
DQA1*0201-DQB1*02
III gruppo
DRB1*03-DQB1*02
DRB1*04-DQB1*0302
DRB1*07-DQB1*02
Questo contributo presenta sinteticamente i dati relativi all’HLA di II classe, DR e DQ e le frequenze aplotipiche:
• nelle popolazioni genericamente “senza celiachia”, non esposte al glutine;
• nelle popolazioni “senza celiachia”, esposte al glutine;
• nelle popolazioni con celiachia a vario grado e presumibilmente predisposte.
Secondo questa indagine, in relazione al presunto ruolo dei suddetti assetti aplotipici DR/DQ, con l’eccezione di “casi unici”, tutte le popolazioni umane sono geneticamente predisposte al morbo celiaco, diretta conseguenza questa della recente evoluzione verso strategie di approvvigionamento diverse dalla caccia e raccolta di frutti spontanei. L’attuale incidenza “nel mondo” è stimata in circa 1/100. Un dato sorprendente: potremmo ipotizzare un eventuale fenomeno di “pandemia cronica” della malattia in seguito alla crescente occidentalizzazione dei modelli alimentari (imposti dal mercato o dalle necessità) e all’uso generalizzato di alimenti contenenti glutine da parte di popolazioni di norma caratterizzate da ecologie diverse.Le popolazioni del Veneto antico( I Paleoveneti ): studio genetico e di coalescenza seriale
Alessandro Manfredini (1), Elise Belle (2), Giovanni Leonardi (3), Angela Ruta Serafini (4), Lucio Milani (5), Francesco Mallegni (1), Guido Barbujani (2), David Caramelli (5).
(1) Dipartimento di Biologia, Università degli Studi di Pisa. (2) Dipartimento di Biologia Evolutiva, Università degli Studi di Ferrara. (3) Dipartimento di Scienze dell’Antichità, Università degli Studi di Padova. (4) Museo Nazionale Atestino, Soprintendenza per i Beni Archeologici del Veneto Padova. (5) Dipartimento di Biologia Animale e Genetica, Laboratori di Antropologia, Università degli Studi di Firenze.
Nel corso degli ultimi anni, studi di paleogenetica su popolazioni dell’Italia antica hanno cercato di ricostruire l’eredità genetica della nostra penisola. Attraverso l’analisi parziale della prima regione di controllo del DNA mitocondriale (HVRI nt 16083-nt16281) recuperato da individui vissuti nel territorio veneto tra l’VIII ed il II secolo a.C. e provenienti da 5 siti archeologici dislocati all’interno della Provincia di Padova, abbiamo cercato di ampliare questo scenario. Nonostante i buoni valori del grado di racemizzazione solo da 17 dei 41 reperti analizzati siamo riusciti ad ottenere una sequenza analizzabile. Al fine di verificare la continuità genetica tra le popolazioni dell’antico veneto e quelle attuali, si è utilizzato un nuovo programma di simulazione seriale, Serial SimCoal (Anderson 2006). In questo modo, tramite simulazioni di coalescenza di modelli genealogici che incorporano le sequenze antiche e moderne, abbiamo indagato vari scenari demografici diversi per taglia delle popolazioni e per tasso di crescita, che hanno potuto portare allo sviluppo della attuale popolazione veneta. Tramite il confronto statistico dei dati di diversità genetica osservata, con quelli simulati è stato possibile escludere alcuni scenari evolutivi, ed evidenziarne altri più probabili.
Linkage Disequilibrium extension analysis on the Xq13 region in the island of Corsica
Veronica Latini (1+), Gabriella Sole (1+), Laurent Varesi (2), Giuseppe Vona (3), Antonio Cao (1) and Maria Serafina Ristaldi(1*)
+ These authors contributed equally to this work. (1) Istituto di Neurogenetica e Neurofarmacologia del Consiglio Nazionale delle Ricerche (INN-CNR), Cagliari, Italy. (2) Universitè de Corte, Corsica, France. (3) Dipt. Biologia Sperimentale, Università di Cagliari.
The identification of genes involved in the pathogenesis of multifactorial diseases would help to shed some light on their physiopathology with significant aid in on the prevention and development of new therapeutic approaches. Genetic isolates with a history of a small founder population, long-lasting isolation and population bottlenecks represent exceptional resources in the identification of disease genes. In these populations the disease allele reveals Linkage Disequilibrium (LD) with markers over significant genetic intervals, therefore facilitating disease locus identification. In a previous work we have examined the LD extension on the Xq13 region in three sub-populations of Corsica belonging to the internal mountainous region of the island. Here we have extended the analysis to the Corsican population of the coast. We found a decreasing of LD in this area. This result indicate a cline of LD inside the island which could be useful for the fine mapping of a gene contributing to a complex disease first mapped using the isolated, high LD, population of the same region. Moreover we reported the frequencies of a particular haplotype (DXS1225-DXS8082) in Corsican population which is typical of the island is not common in other European populations.
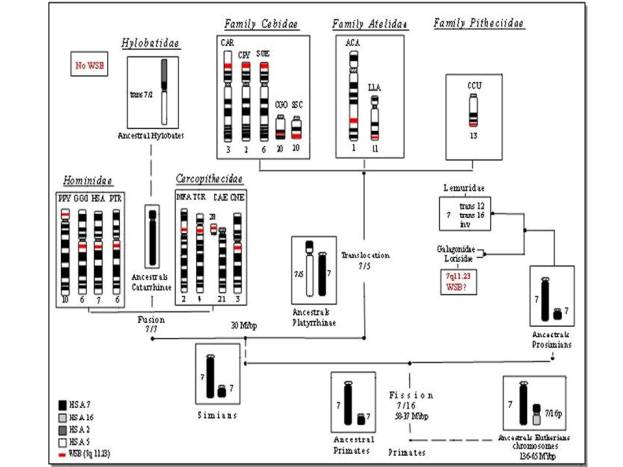
Localizzazione cromosomica ed analisi delle dinamiche evolutive del locus HSA7q11.23 “William-Beuren” nei primati
Barbara Picone(1*), Francesca Dumas(1), Roscoe Stanyon(2), Antonella Lannino(1), Roberto Vitturi(1), Francesca Bigoni(2), Orsola Privitera(3), Daniela Romagno(1), Luca Sineo(1).
(1) Dipartimento di Biologia animale “G. Reverberi” – Università degli Studi di Palermo – b.picone@unipa.it / * Department of Botany and Zoology-Evolutionary Genomics Group - University of Stellenbosch, Cape Province - South Africa / (2) Dipartimento di Biologia animale e Genetica – Università degli Studi di Firenze / (3) Laboratorio di Citogenetica – Unità Ospedaliera Legnano - Milano
La mappatura di loci specifici di dimensioni ridotte (800-450 Kb) attribuisce un notevole potere risolutivo allo studio dei riarrangiamenti cromosomici di interesse filogenetico. Il locus conosciuto come Williams-Beuren, dal nome della sindrome umana da microdelezione, mappa nel braccio q del cromosoma 7. La sintenia 7 ha un’origine molto complessa, investigata e descritta di recente (Murphy et al., 2001; Müller et al., 2004). La sintenia, nella sua evoluzione nei mammiferi e in particolare nei primati, è riscontrabile sotto forma di due distinti segmenti, uno dei quali, di minor dimensioni, si ritrova frequentemente associato con altre sintenie, quali la #16 (Euteri “ancestrali” e Strepsirrhini) e la #5 (Platyrrhini). Metafasi di H.sapiens, P.pygmaeus, P.troglodytes, G.gorilla, C.aethiops, C.neglectus, M.fascicularis, T.cristata, C.argentata (M.argentatus), C.cupreus, A.caraja, S.oedipus, C.goeldii, S.sciureus, A.paniscus, L.lagothrica, C.pygmaea sono state ibridate con le metodiche correnti. Con l’eccezione di HSA, PPY, GGO, PTR, CAE, MFA e TCR (LS e DR) tutti i campioni sono stati preparati da RS presso i laboratori del National Cancer Institute (Frederick, USA). Le ricerche effettuate nel periodo 2003-2007 (in parte pubblicate: Sineo e Romagno 2006; Sineo et al., 2007) hanno portato: al mappaggio, nelle diverse specie di Anthropoidea, del locus HSA7q11.23, che si mostra, con un’unica eccezione (CCU), incluso nella sintenia comprendente il frammento HSA 7p22-qter - 7q 22.22, e a valutazioni circa l’evoluzione della sintenia e le relazioni tra le specie. Questo approccio si è dimostrato utile nell’indagine di riarrangiamenti cromosomici individuati con il banding e quale supporto essenziale all’analisi genomica condotta mediante le metodologie di Reciprocal Painting.
Müller S, Finelli A, Neusser M, Wienberg J. 2004. The evolutionary History of Chromosome 7. Genomics 84: 458-467
Murphy WJ, Stanyon R, O’Brien S. 2001. Evolution of Mammalian genome organization inferred from comparative gene mapping. Genomic Biology 2 (6: Rev. 0005.1-.8)
Sineo L e Romagno D. 2006. Born and Rise of Human Chromosome 7 Syntenies. In: Sineo L, Stanyon R (eds) Primate Cytogenetics and Comparative Genomics. Firenze Firenze University Press, pp. 57-66
Sineo L, Dumas F, Vitturi R, Picone B, Privitera O, Stanyon R. 2007. William Beuren mapping in C. argentata, C. cupreus and Alouatta caraja indicates different patterns of chromosomal rearrangements in neotropical primates. J Zool Syst Evol Res Published article online: 14-May-2007
Museum Primatological Collections: a valuable source of ancient DNA
Giulio Catalano (1), David Caramelli (1), Luca Sineo (2)
(1) Dipartimento di biologia animale e Genetica “Leo Pardi”, Università di Firenze, Italia (2) Dipartimento di Biologia animale “Giuseppe Reverberi”, Università degli Studi di Palermo, Italia
Recent advances in DNA technology have made possible to recover DNA from archaeological and palaeontological remains allowing to the scientist to go back in time studying the genetic relationships of extinct organisms to their contemporary relatives. In particular mammals museum collections represents a good source of materials that can be very important in the light of systematics and phylogenetic surveys as they can be representative of an extinct population or of an ancient genetic variability. One of the new and most important field of application ancient molecular Primatology (AMP). Due to the ecological pressure and progressive deforestation extant wild ranging primates are a very limited representation of ancient wild populations. Several extinction events took place in recent decades. Issues in systematics and even more in phylogeny are still open as like a series of interesting questions on the evolution of gene sequences as like lactase, alcohol dehydrogenase, or T-cell receptor architecture. Here we present preliminary results obtained by DNA extraction, PCR specific-amplification, cloning and sequencing of mitochondrial 16S ribosomal RNA from museum bone specimens of Pongo pygmaeus (Primates, Catarrhinae, Pongidae).
Polimorfismo del gene ACTN3 e ginnasti d'elite
Myosotis Massidda (1) (2)
(1) Dipartimento di Biologia Evoluzionistica e Sperimentale, Facoltà di Scienze Motorie, Università di Bologna (2) Corso di Laurea in Scienze Motorie, Università di Cagliari
Le prove scientifiche a favore delle influenze genetiche sulla prestazione sportiva d’elite hanno subito un forte incremento nell’ultimo decennio. Differenze significative nella distribuzione delle frequenze del genotipo dell’ACTN3 (alfa-actinina 3) sono state riscontrate tra gli atleti praticanti discipline di sprint/potenza e di endurance.
Il polimorfismo del gene ACTN3 si presenta con due varianti alleliche (R e X) e quindi 3 diversi genotipi: 577RR, 577XX e R577X.
L’allele 577R del gene ACTN3 codifica per la produzione della proteina alfa-actinina 3, che è espressa esclusivamente nel disco Z delle fibre di tipo II del muscolo scheletrico, responsabili della generazione di forti contrazioni ad alte velocità. L’alfa-actinina 3 gioca un ruolo decisivo nel legame con i filamenti dell’actina assumendo sia una funzione statica nell'effettuare l'allineamento miofibrillare, sia una funzione regolatrice nella contrazione muscolare.
Recenti risultati sugli atleti d’elite suggeriscono che l’allele 577R conferisce un vantaggio nella performance di sprint e potenza ad alti livelli agonistici e lo scopo del presente lavoro è quello di verificare se tale vantaggio sussiste anche nella performance sportiva d’elite della ginnastica artistica.
Per verificare questa ipotesi, abbiamo studiato la distribuzione allelica e genotipica dell’ACTN3 sui 35 Ginnasti delle Squadre Nazionali Italiane (2007) Juniores e Seniores di Ginnastica Artistica (17 maschi e 18 femmine) che avevano raggiunti livelli agonistici Mondiali ed Olimpici. I risultati ottenuti sono stati confrontati con un gruppo di controllo formato da 53 soggetti (31 maschi e 22 femmine) sani e sedentari. Il DNA è stato estratto da ciascun soggetto utilizzando un tampone salivare. L’elaborazione statistica dei dati è stata condotta mediante il programma Genepop (v.4.0.3).
Differenze significative (p=0.035) nella distribuzione delle frequenze genotipiche si sono riscontrate tra gruppo dei ginnasti: RR=48.57%; RX=48.57% e XX=2.86% e gruppo di controllo: RR=32.07%; RX=49.05%; XX=18.86%. Si sono evidenziate frequenze significativamente più alte dell’allele 577R nei ginnasti rispetto al controllo (R=72.85% vs R=56.60%). I nostri risultati si trovano in accordo con quanto descritto in letteratura relativamente all’associazione tra polimorfismo dell’ACTN3 e performance sportiva degli atleti di sprint (Yang et al., 2003). Questi ultimi, infatti, presentano frequenze significativamente più elevate dell’allele 577R rispetto alla popolazione normale ed agli atleti di endurance. Ciò suggerisce che la presenza di alfa-actinina-3 abbia un effetto benefico sulla funzione del muscolo scheletrico nella generazione di forti contrazioni ad alte velocità. In considerazione dei risultati ottenuti e analizzata la natura della performance nella ginnastica artistica (caratterizzata dall’esecuzione di esercizi acrobatici che richiedono lo sviluppo di forti contrazioni muscolari ad alte velocità), si può concludere che il ruolo assunto dall’alfa-actinina 3 nel favorire la performance d’elite di sprint e potenza (Yang. et al., 2003; Niemi e Majamaa, 2005) possa essere il medesimo anche per la ginnastica artistica.Prevalenza dei fattori di rischio delle malattie cardiovascolari nella popolazione Corsa.
Alessandra Falchi (1), Ignazio S Piras (2), Carla M Calo’ (2), Pedro Moral (3), Giuseppe Vona (2), Laurent Varesi (1)
(1) Department of Human Genetics, University of Corsica (France). (2) Department of Experimental Biology, University of Cagliari (Italy). (3) Department of Animal Biology, Section of Anthropology, University of Barcelona (Spain).
In questo studio, sono stati analizzati sette polimorfismi in 100 individui presentanti una sintomatologia tipica delle coronopatie e in 100 individui sani originari del centro della Corsica (Francia).
Questi sette polimorfismi sono localizzati in sei regioni geniche coinvolte: 1) Regolazione del sistema renina-angiotensina (ACE I/D), 2) Metabolismo dei lipidi: Colesterolo ester transferasi (CETP TAQ1B), 3) Aggregazione delle piastrine: subunità alfa e beta del complesso delle integrine (GpIIb HPA3 and GpIIIa Pl A1/A2), 4) Fibrinolisi: Attivatore del Plasminogeno (PLAT TPA25 I/D), Metilenetraidrofolato reduttasi (MTHFR C677T e A1298C).
I campioni sono stati amplificati e digeriti tramite gli enzimi di restrizione (RFLPs).
Nessuna differenza significativa è stata trovata tra il gruppo dei pazienti e dei controlli per quanto riguarda le frequenze alleliche.
La frequenza del genotipo MTHFR T677T e dell’aplotipo T677T /A1298A è più elevata nei casi (20%) che nei controlli (4%). L’Odds ratio sembrerebbe indicare che gli individui che presentano il genotipo MTHFR T677T e l’aplotipo T677T/A1298A presentano un rischio sei volte superiore di sviluppare la malattia cardiovascolare (OR= 6; 95% CI =1.96-18.28) suggerendo una possibile associazione del genotipo MTHFR T677T con il rischio di coronoropatie nella popolazione corsa.
Questa ricerca è stata realizzata grazie ai fondi della comunità europea (Programmes INTERREG).Progetto Yanesha: analisi della variabilità genetico-molecolare di una popolazione della Selva Centrale peruviana
Giorgio Piracci, Daniele Yang Yao, Chiara Barbieri, Graziella Ciani, Loredana Castrì, Donata Luiselli, Davide Pettener
Dipartimento di Biologia E.S., Area di Antropologia, Università di Bologna
La presente ricerca si propone di analizzare la struttura genetica, individuare le origini di un gruppo nativo amerindiano della Selva Centrale del Perù e stimarne il grado di admixture in seguito ai fenomeni migratori e di colonizzazione subiti.
Gli Yanesha, sino ad ora studiati solo su base linguistica, si sarebbero distaccati dal gruppo madre Arawak circa 4000 anni fa, andando incontro a progressivi mescolamenti con le popolazioni Quechua dell’area subandina. La popolazione Yanesha è attualmente costituita da poco meno di 10.000 individui, distribuiti in 31 comunità stanziali della provincia di Oxapampa. Le comunità sono disperse in tre macro aree con diverse caratteristiche ambientali e socioeconomiche: zona alta (700- 2000 m.s.l.m.), zona media (300- 700 m.s.l.m.) e zona bassa (<300 m). Le tre macro aree hanno anche un significato storico-demografico: la parte alta rappresenta infatti l’antico areale originario, precedente alla colonizzazione spagnola, mentre le altre due costituiscono le zone popolate tra il 1530 ed oggi in seguito alla pressione colonizzatrice.
Nel corso di una spedizione effettuata nel maggio 2007 sono stati raccolti oltre 300 campioni di mucosa buccale provenienti da individui di 6 comunità della selva alta (Tsachopen, Mayme, Ñagazu, Milagros, Cacazù, Yurinaqui) e 5 della selva media (Nueva Esperanza, 7 de Junio, Alto Isco, Shiringa, Loma Linda). Per ciascun individuo è disponibile una scheda biodemografica con informazioni su cognomi, etnia, nascita e residenza dei genitori e nonni dei soggetti campionati. Il campionamento è stato effettuato con il consenso informato dei singoli soggetti ed a seguito di un protocollo di intesa firmato con il Cornesha delle Comunità.
La presente ricerca si propone dunque di analizzare la variabilità genetica a livello mitocondriale (HVRI e RFLPs della regione codificante) e del cromosoma Y (SNPs e STRs) di una popolazione di origine amazzonica che può costituire, per la sua localizzazione geografica e la sua storia, lo strumento per inferire alcuni dei principali movimenti migratori che hanno interessato il sud-America.
I risultati preliminari fino ad ora ottenuti sono finalizzati ad una serie di obiettivi specifici: 1. prima caratterizzazione genetico-molecolare di una popolazione di origine Arawak; 2. analisi della distribuzione della variabilità dei marcatori di linea; 3. analisi dei patterns di flusso genico e di deriva che hanno interessato le singole comunità; 4. stima dei livelli di admixture con individuazione dei contributo genetici derivanti dal contatto con i gruppi Quechua Andini e con popolazioni europee.
Quanto incidono le contaminazioni umane moderne sui reperti ossei antichi?
Elena Pilli (1), Martina Lari (1), Lucio Milani (1), Alessandra Modi (1), Giulio Catalano(1), Barbara Lippi (2), Francesca Bertoldi (2), Sauro Gelichi (3), Francesco Mallegni (2), David Caramelli (1)
(1) Laboratori di Antropologia, Dipartimento di Biologia Animale e Genetica, Università degli Studi di Firenze (2) Dipartimento di Biologia, Università di Pisa (3) Dipartimento di Scienze dell'Antichità e del Vicino Oriente, Università Cà Foscari, Venezia
Gli studi sul DNA umano antico utilizzano principalmente ossa e denti come fonti di materiale genetico. Nel corso delle loro storia tafonomica questi resti umani vanno inevitabilmente incontro a contaminazioni sia da parte di microrganismi che, crescendo saprofiticamente a spese del materiale cellulare, lo degradano e lo impoveriscono del DNA originale, sia da parte della manipolazione del resto stesso compiuta dagli operatori che maneggiano i campioni. Nonostante l’attenzione del mondo scientifico nello sviluppo di sistemi atti alla rimozione anche meccanica di eventuali contaminanti presenti e nell’elaborazione di sofisticati metodi di autenticazione dei risultati ottenuti con l’analisi del DNA antico, il problema delle contaminazioni dei campioni ossei rimane uno dei problemi fondamentali e non ancora largamente. Tutto ciò incide molto sull’attendibilità dei risultati ottenuti. In questo lavoro, allo scopo di studiare e capire gli effetti della manipolazione sull’ottenimento del profilo genetico autentico del campione, abbiamo deciso di analizzare e confrontare i profili mitocondriali ottenuti in seguito all’analisi di distretti ossei diversi (femore, ulna, costa e dente) appartenenti ad individui diversi organizzati in due set di campioni: campioni “vergini” cioè prelevati dagli archeologi con guanti, mascherina e camice e immediatamente mandati in laboratorio per l’analisi e campioni “lab” manipolati dagli studiosi come sono soliti fare. Lo scopo del lavoro è stato quello di investigare:
i) quanto i metodi di pulizia applicati nell’analisi del DNA antico siano atti alla rimozione delle contaminazioni dovute a DNA moderno,
ii) quanto la manipolazione del campione da parte degli operatori possa incidere sull’ottenimento di un profilo mitocondriale autentico, agendo in qualche modo sulla qualità e/o sulla quantità del materiale genetico presente,
iii) se esistano distretti ossei maggiormente soggetti alle contaminazioni.
Al fine di poter escludere la presenza di eventuali profili contaminanti esogeni dovuti alla manipolazione, tutte le persone che hanno scavato ed analizzato i campioni sono state tipizzate in modo da poter sostenere l’autenticità del dato genetico ottenuto. I risultati preliminari ottenuti da questa analisi hanno mostrato, in primo luogo, come sia stato più facile ottenere un primo profilo genetico parziale dai campioni “vergini” rispetto ai campioni “lab.” facendo questo ipotizzare che il DNA nei campioni “vergini”, non manipolati, sia in qualche modo meglio conservato e/o in quantità maggiore, condizione quest’ultima presumibilmente dovuta alla manipolazione ed al lavaggio a cui i campioni vengono sottoposti da parte degli archeologi. I dati inoltre sembrano sostenere l’efficacia dei metodi di pulizia applicati: non sono state infatti fino ad ora riscontrate sequenze contaminanti riconducibili a coloro che hanno manipolato le ossa. Infine ancora non è emerso alcun dato chiaro che possa evidenziare se ci sia un distretto osseo più suscettibile alle contaminazioni.Studio della variabilità genetica di un campione della popolazione della Basilicata
Luciana Vitelli (1), Claudio Ottoni (2), Cristina Martinez Labarga (2), Olga Rickards (2)
(1) Dipartimento di Biologia Animale e Genetica, Università degli Studi di Firenze (2) Dipartimento di Biologia, Università degli Studi di Tor Vergata, Roma
La Basilicata, grazie alla sua posizione geografica nel cuore dell’Italia meridionale, ai due sbocchi sul mare, uno sul versante ionico e l’atro sul versante tirrenico, e alla ricchezza di fiumi che rendono il suo territorio particolarmente fertile, è sempre stata una terra contesa e un importante crocevia di popoli e culture, che in ogni epoca hanno lasciato le testimonianze del loro passaggio.
Per questo lavoro è stato preso in esame un campione di 80 individui di accertata genealogia materna lucana. L’estrazione del DNA è stata effettuata da mucosa buccale. È stata studiata la variabilità genetica a livello del mtDNA: per ciascun individuo sono state ottenute le sequenze dei segmenti ipervariabili HVS-I (15996-16401) e HVS-II (00029-00408) del D-loop; inoltre sono stati analizzati, tramite enzimi di restrizione, alcuni polimorfismi della regione codificante particolarmente informativi per la classificazione.
All’interno del campione sono stati individuati 58 aplotipi diversi, classificabili in 13 aplogruppi. I risultati osservati sono stati confrontati con quelli già noti per l’Italia centro-meridionale e con quelli di altre popolazioni dell’Europa e del bacino del Mediterraneo, pubblicati in letteratura.
Dal confronto della distribuzione delle frequenze degli aplogruppi in Basilicata e nell’Italia centrale la popolazione in esame rientra chiaramente nella variabilità caratteristica della nostra penisola. La differenza più evidente rispetto alle altre regioni è l’elevata frequenza dell’aplogruppo K.
Il pattern di variabilità osservato a livello delle sequenze si inserisce nel contesto filogenetico dell’area del Mediterraneo.
In conclusione, i dati presentati sul campione lucano sono concordi con tutti quelli già noti, confermando quanto inferito dalle ricostruzioni paleoantropologiche e archeologiche.Studio della variabilità mitocondriale delle popolazioni del Bird’s Head (Nuova Guinea indonesiana)
Immacolata Cascione (1), Marcella Attimonelli (2), Yoni Syukriani (3), Saifuddin A Noer (3), Herawati Sudoyo (4), Sangkot Marzuki (4), Mila Tommaseo Ponzetta (5)
(1) Dipartimento di Genetica e Microbiologia, Università di Bari (2)Dipartimento di Biochimica e Biologia molecolare, Università di Bari (3) PPAU Bioteknologi, Institut Teknologi Bandung, Bandung, Indonesia (4) Eijkman Institute for Molecular Biology, Jakarta, Indonesia (5) Dipartimento di Zoologia, Università di Bari
La Penisola del Bird’s Head costituisce l’estremità nord-occidentale della Nuova Guinea e si configura come un’area di transizione culturale e linguistica tra la parte orientale dell’Indonesia insulare e la vicina Melanesia. Fin dal primo popolamento della Nuova Guinea questa penisola è stata porta d’ingresso di migrazioni umane e di successivi contatti e scambi con i vicini arcipelaghi, le cui tracce sono evidenti nella varietà culturale e linguistica delle sue popolazioni, parlanti sia lingue Papua che Austronesiane.
Sono qui presentati i primi risultati di un’indagine sulla variabilità mitocondriale delle popolazioni Bird’s Head e della Nuova Guinea nord-occidentale. È stata analizzata la regione ipervariabile 1 (HVS1) del DNA mitocondriale estratto da radice di capello e/o da cellule dell’epitelio della mucosa buccale di 265 soggetti appartenenti a 15 gruppi etnici diversi. Il sequenziamento ha identificato 90 siti varianti, che definiscono 108 aplotipi, di cui 46 sono condivisi. È stata inoltre effettuata l’analisi RFLP della regione codificante. Le due analisi congiunte hanno reso possibile l’attribuzione dei soggetti a 13 sottoaplogruppi appartenenti ai due maggiori raggruppamenti M ed N.
La diversità aplotipica e nucleotidica all’interno delle popolazioni è stata stimata mediante il calcolo di FST e AMOVA. Un albero Neighbor-Joining senza radice, costruito utilizzando la matrice delle distanze FST, e un Median Joining Network, ottenuto mediante il programma NETWORK, hanno permesso di evidenziare le relazioni degli aplotipi all’interno di ciascun aplogruppo. Lo stesso programma è stato utilizzato per calcolare il tempo di coalescenza di ogni aplogruppo, successivamente confrontato con quelli riportati in letteratura.
I risultati confermano l’alta eterogeneità popolazionistica e la condivisione di aplotipi tra gruppi, a prescindere dalla diversità linguistica e dalla loro collocazione geografica.
La frequenza dell’aplogruppo Q1 prevale nelle popolazioni del versante sud occidentale della regione, raggiungendo il 90% negli Ekkari, confermando così una rete di collegamenti e contatti con la parte centrale dell’isola, dove questo aplogruppo è prevalente. Gli aplogruppi P1 e B sono variamente distribuiti e relativamente frequenti negli abitanti dell’area costiera orientale e delle isole. Le datazioni degli aplogruppi ne indicano l’antichità, e – come già ipotizzato da Birdsell (1977) - confermano questa penisola come punto d’approdo privilegiato per il primo popolamento del Sahul, il paleocontinente che nel Pleistocene riuniva Nuova Guinea e Australia.
Birdsell JB, 1977. The recalibration of a paradigm for the first peopling of Greater Australia. In: J.Allen, J.Golson and R.Jones (eds.), Sunda and Sahul: Prehistoric Studies in Southeast Asia, Melanesia, and Australia, pp. 113-168, New York, Academic Press.The role of geography and language in the shaping of Y genetic variability in the Caucasus
Laura Caciagli (1), Sergio Tofanelli (1), Kazima Bulayeva (2)(3), Luca Taglioli (1), Giorgio Paoli(1)
(1) Dipartimento di Biologia, Università di Pisa, Italy (2) Vavilov Institute of General Genetics, Russian Academy of Sciences, Moscow, Russia (3) Institute of History, Archeology and Ethnology, Daghestan Center of the Russian Academy of Sciences, Makhachkala, Daghestan, Russia
The Caucasus region provides a great opportunity to investigate the influence of geography and language on the genetic structure of human populations. About 50 ethnic groups speaking Caucasian, Indo-European and Turkic languages live in this region, while mountain ranges reaching over 5000 m represent strong geographic barriers to gene flows.
In order to investigate the genetic variability and relationships of Caucasus populations, 17 YSTR loci were analyzed among seven ethnic groups. from Daghestan (Northern Caucasus)..
Genomic DNA from unrelated informed donors (N=144) was PCR-amplified using the "AmpFlSTR®Y-filerT" kit (Applied Biosystems®). The length of PCR products was estimated with the ABI-PRISM 310 genetic analyzer using the GeneScan® (Applied Biosystems®) software. The assignment of YSTR haplotypes to binary states defining haplogroups was obtained by a Bayesian method (Athey, 2006) with output scores > 70. The basic parameters of molecular diversity and population genetic structure, including the concordance between geography, language and genetic structure was investigated by AMOVA and Mantel tests using the software Arlequin 3.0.
The results showed that the seven Daghestan ethnic groups fall into the range of Y-STR and Y-SNP variability of Caucasian populations, However some peculiar characteristics make Daghestan region an outlier in the Caucasus genetic scene. The mean level of genetic differentiation (FST = 0.138; p = 0.0000) was high and significant. Private alleles were found at DYS458, DYS448 and DYS385 loci.
Linguistic barriers didn’t appear the main determinants of the high between-population Y-chromosome variability observed in the Caucasus. Eventually, founder effect and long-term genetic drift caused by the rigid structuring of societies in groups of patrilineal descent remain the best explanation of the genetic divergence among Caucasian ethnics and of the presence of rare allelic variants.
Athey WT 2006. Haplogroup Prediction from Y-STR Values Using a Bayesian-Allele-Frequency Approach. J of Genet Geneal 2:34-39.Tipizzazione del locus diagnostico G37995C del gene nucleare della microcefalina in un reperto neandertaliano italiano
Lucio Milani (1), Martina Lari (1), Giulio Catalano (1), Elena Pilli (1), Alessandro Manfredini (1), Laura Longo (2), Silvana Condemi (3), David Caramelli (1).
(1) Laboratori di Antropologia, Dipartimento di Biologia Animale e Genetica, Università di Firenze (2) Dipartimento di Scienze Ambientali, Università di Siena (3) CNRS, UMR 6578: “Unité d’Antropologie: Adaptabilité Biologique et Culturelle“. Faculté de Médecine – Univeristé de la Méditerranée, Marseille, France.
Nel dibattito che riguarda l’origine dell’uomo moderno e i suoi rapporti evolutivi con le forme arcaiche di Homo, in particolare i Neandertaliani, resta ancora da chiarire se tra le due specie ci sia stata ibridazione e se questi ultimi abbiano in qualche modo contribuito alla costituzione del pool genetico moderno. La difficoltà di valutare l’entità di questo contributo risiede nel fatto che, a causa della deriva genetica, bassi livelli di flusso genico, come quello che può essere avvenuto dai Neandertaliani verso i primi Homo sapiens, non hanno lasciato grosse tracce nel pool genetico moderno. L’unico modo per poter identificare nei nostri geni un eventuale contributo dei Neandertaliani è legato all’introgressione di alleli sottoposti a selezione positiva. Recentemente è stato individuato un marcatore, il gene della microcefalina, una proteina che interviene durante lo sviluppo del sistema nervoso, che è stato sottoposto a selezione positiva lungo la linea evolutiva umana. Particolare attenzione è stata rivolta alla posizione 37995 in cui la presenza di una citosina (allele derivato, D) al posto della guanina, mutazione che determina un cambiamento aminoacidico nella proteina, caratterizza una serie di aplotipi nettamente divergenti dagli altri; l’allele D è di origine recente (≈37.000 anni fa) ma è presente attualmente ad alta frequenza in particolare nelle popolazioni europee (>70%). Per spiegare questo particolare andamento al locus MCPH1, tramite analisi statistiche di coalescenza seriale è stato ipotizzato che l’allele D rappresenti un esempio di flusso genico dalla popolazione neandertaliana, in cui esso era fissato, verso la popolazione degli antichi sapiens, in cui invece tale allele era assente; questo evento di ibridazione sarebbe stato raro ed avrebbe portato all’introgressione di una singola copia dell’allele che poi si sarebbe diffuso sotto la spinta della selezione positiva nelle popolazioni sapiens fino a raggiungere le frequenze attuali.
Nonostante le difficoltà che caratterizzano il recupero e l’analisi del DNA nucleare da reperti antichi, per verificare questa ipotesi abbiamo amplificato, clonato e sequenziato un piccolo frammento di circa 70 paia basi attorno alla posizione 37995 del gene MCPH1 di un reperto neandertaliano italiano di circa 50.000 anni fa proveniente da Riparo Mezzena, nei monti Lessini (VR); sulla base dei risultati ottenuti da una serie di analisi precedenti tale reperto si era rivelato, infatti, eccezionalmente ben preservato e privo di contaminazioni da parte di DNA umano moderno.
Abbiamo riscontrato che tutte le sequenze di MCPH1 ottenute presentano alla posizione 37995 l’allele ancestrale G. Alla luce di questo dato, se confermato, sembra improbabile che l’allele D sia il retaggio di un flusso genico avvenuto dal pool nenadertaliano verso quello sapiens, mentre è più probabile che tale allele si sia originato per mutazione nella linea evolutiva di Homo sapiens.Valutazione della pressione selettiva in 17 STRs nelle popolazioni del Mediterraneo
Ignazio Piras (1,2), Alessandra Falchi (2), Pedro Moral (3), Giorgio Paoli (4), Elena Ghiani (1), Alessandra Melis (1,2), Carla Calò (1), Laurent Varesi (2), Giuseppe Vona (1)
(1) Dipartimento di Biologia Sperimentale – Università di Cagliari (2) Department de Génétique Moleculaire – Université de Corse (3) Departament de Biologia Animal – Universitat de Barcelona (4) Dipartimento di Etologia, Ecologia, Evoluzione – Università di Pisa
L’individuazione dei loci che hanno subito una recente pressione selettiva è utile sia da un punto di vista epidemiogenetico che antropologico molecolare. È possibile identificare markers correlati con la differente suscettibilità alle patologie e individuare i marcatori non “neutri”, non utilizzabili nelle analisi popolazionistiche. Sono stati elaborati numerosi test di neutralità selettiva, basati su vari modelli di genetica di popolazione. Tale caratteristica rende necessario l’utilizzo simultaneo di più test, identificando come loci candidati ad aver subito fenomeni di selezione naturale esclusivamente quelli che risultano significativi in tutti i test.
Lo scopo di questo lavoro è stato quello di individuare dei loci la cui variabilità è stata influenzata dalla pressione selettiva in alcune popolazioni umane del Mediterraneo.
Il campione analizzato consiste di 429 individui provenienti da 6 popolazioni del Mediterraneo (Sardegna, Spagna, Isole Baleari, Sicilia, Toscana e Marocco). I polimorfismi analizzati (17 STRs) sono localizzati in geni codificanti per enzimi del metabolismo ossidativo, proteine del sistema immunitario ed eritrocitarie. L’analisi è stata eseguita mediante PCR con primers fluorescenti e lettura con ABI 3730 DNA analyzer. I dati sono stati elaborati applicando tre test di neutralità selettiva basati sui confronti a coppie tra le popolazioni: Fst, F e LnRH test (Beaumont and Nichols, 1996; Vitalis et al., 2001; Schlotterer, 2002).
I risultati mostrano il 15.6% di scostamenti dai modelli di neutralità (il 67.3% con significatività all’1%). Confrontando i tre test applicati, l’F test mostra il 34.4% dei confronti significativi, rispetto al 6.7% e 5.9% dell’Fst e LnRH test rispettivamente. Due polimorfismi mostrano scostamento dalla neutralità in tutti i test: il TNFe (GA)n (Sicilia vs Toscana), il NOS1 EX29 (Sicilia vs Marocco e Isole Baleari vs Marocco).
In conclusione, i possibili loci soggetti a una selezione naturale o in Linkage Disequilibrium con una regione che presenti questi patterns sono il TNFe (GA)n e il NOS1 (CA)n EX29. I risultati non dimostrano la presenza della selezione, ma permettono di identificare loci sui quali svolgere valutazioni più dettagliate. La non concordanza dei risultati ottenuti con i differenti test, già osservata in altri lavori, è probabilmente dovuta alla difficoltà di valutare la storia demografica delle popolazioni e il tasso di mutazione dei loci, che porta alla mancata assunzione di tutte le condizioni previste dai modelli utilizzati nei test di neutralità.
Bibliografia
Beaumont MA, and Nichols RA. Evaluating loci for use in the genetic analysis of population structure. 1996. Proc R Soc Lon 263:1619-1626.
Schlotterer C. 2002. A microsatellite-based multilocus screen for the identification of local selective sweeps. Genetics 160:753-763.
Vitalis R, Dawson K, and Boursot P. 2001. Interpretation of variation across marker loci as evidence of selection. Genetics 158:1811-1823Variabilità del gene TNFRSF13B (TACI) in campioni di popolazioni italiane e africane
Marco Sazzini (1), Roberta Zuntini (2), Simona Ferrari (2), Giovanni Romeo (2), Donata Luiselli (1)
(1) Lab. di Antropologia molecolare, Dipartimento di Biologia Evoluzionistica Sperimentale (BES), Università di Bologna - (2) Azienda Ospedaliero-Universitaria S.Orsola-Malpighi –U.O. Genetica Medica - Bologna
L’Immunodeficienza Comune Variabile (CVID) è una patologia che presenta un quadro clinico complesso comprendente disordini a carico del sistema immunitario caratterizzati da una ridotta risposta anticorpale dovuta a bassi livelli di immunoglobuline nel siero.
Mutazioni a carico del gene TNFRSF13B, in posizione 17p11.2 e codificante per TACI, risultano associate a CVID o a deficit di IgA.
TACI è infatti un Tumor Necrosis Factor Receptor che viene espresso sulla superficie dei linfociti B regolandone la sopravvivenza, lo switching isotipico e la risposta anticorpale mediante l’interazione con specifici ligandi.
Lo scopo del presente lavoro è stato quello di condurre un’analisi della diversità aplotipica in soggetti italiani malati e sani, per valutare se vi fosse una differenza di associazioni fra mutazioni causative e combinazioni di stati allelici nei differenti gruppi. È stato inoltre esaminato un pannello di campioni africani al fine di ampliare il quadro già esistente della variabilità del gene, mai studiato prima in popolazioni extra-europee.
I 5 esoni costituenti TNFRSF13B sono stati analizzati mediante sequenziamento diretto in 115 individui affetti da CVID, 96 controlli sani rappresentativi della popolazione italiana, 96 controlli sani provenienti dalla Val Scalve (BG) e 96 controlli sani rappresentativi della maggior parte del continente africano.
Sono stati identificati 21 polimorfismi e 7 mutazioni causative. Quattro siti variabili riscontrati nel campione africano non sono mai stati riportati prima in letteratura.
Sono stati inferiti 51 aplotipi di cui 21 risultano condivisi da due o più gruppi.
La matrice di distanze genetiche, calcolata mediante la stima delle differenze a coppie fra i vari gruppi, ha dato valori significativi in tutti i confronti riguardanti il campione africano e il campione della Val Scalve (p=0.000). Sono invece risultate non significative le distanze fra soggetti malati e controlli sani (p=0.82).
Il confronto effettuato considerando tre gruppi principali (malati, sani, africani) mediante analisi della varianza molecolare (AMOVA) ha evidenziato una valore di variabilità tra gruppi del 2,09 % (FCT=0,02087; p=0,53), dell'11,93 % fra le popolazioni entro lo stesso gruppo e del 85,98 % fra gli individui entro popolazione. Il campione della Val Scalve ha mostrato i valori minori per quanto concerne il numero di siti polimorfici (S=8), gli aplotipi inferiti (K=11) e la diversità aplotipica (H=0,694 ± 0,026). Inoltre il suo aplotipo a maggiore frequenza (48 %), è risultato essere specifico del gruppo e privo di alleli mutati. Pur ipotizzando che si sia verificato un effetto fondatore e che la popolazione scalvina sia andata incontro ad un forte isolamento, tale aplotipo non è stato trovato nel campione rappresentativo della popolazione italiana.
Interessante è infine la totale assenza di mutazioni causative nel campione africano che potrebbe rappresentare una conferma dell’impatto che esse hanno sullo sviluppo della patologia, se fosse vera l’ipotesi che in paesi privi di una copertura sanitaria adeguata la sopravvivenza degli individui affetti da CVID risulta fortemente compromessa.Variabilità genetica a livello del DNA mitocondriale nei balcani
Cristina Martínez-Labarga (1), Romina Mosticone (1), Gabriele Scorrano (1), Irene Contini (1), Gianfranco Biondi (2), Olga Rickards (1)
(1) Centro di Antropologia molecolare per lo studio del DNA antico, Dipartimento di Biologia, Università di Roma “Tor Vergata”, Via della Ricerca Scientifica n. 1, 00173 Roma, Italia. FAX: 06-2023500 Telefono: 06-72594348 Mail: cristina.martinez@uniroma2.it (2) Dipartimento di Scienze Ambientali, Università dell’Aquila, Via Vetoio s.n.c., 67100 Coppito di L’Aquila, Italia.
La storia umana nei Balcani è molto complessa a causa dei numerosi processi di migrazione che hanno interessato la penisola già dalla prima occupazione da parte dell’umanità anatomicamente moderna, e della presenza di popoli che parlano lingue appartenenti a diverse sottofamiglie della famiglia linguistica Indo-Europea.
In questa ricerca abbiamo analizzato la variabilità genetica, a livello del DNA mitocondriale (mtDNA), in un campione di individui di origine serba e per avere una visione complessiva della diversità genetica nei Balcani, abbiamo confrontato le sequenze ottenute con i dati, raccolti in letteratura, che riguardano le altre popolazioni dell’area.
Il DNA genomico è stato estratto da mucosa buccale di 54 adulti. Sono state sequenziate le due regioni ipervariabili del D-loop e analizzate alcune posizioni polimorfiche della zona codificante, diagnostiche per l’inserimento dei vari aplotipi nella filogenesi mondiale dell’mtDNA proposta da Macaulay and Richards (http://www.stats.gla.ac.uk/~vincent/images/skeleton07-08-02.jpg).
A partire dalle sequenze dei due segmenti del D-loop sono stati analizzati alcuni indici di diversità e sono state effettuate delle analisi statistiche che hanno permesso di estrarre informazioni di carattere genetico e demografico. Le sequenze, i cui rapporti sono stati delineati attraverso la costruzione di un Network, sono state attribuite a 16 aplogruppi diversi, 15 di origine eurasiatica ed uno solo di origine africana. I valori di divergenza genetica hanno messo in evidenza un elevato grado di variabilità genetica mentre la rappresentazione della distribuzione delle differenze nucleotidiche tra coppie di sequenze, e il valore del test di neutralità selettiva di Tajima suggeriscono che la popolazione sia andata incontro ad una recente espansione demografica.
L’analisi delle sequenze dei campioni dell’area dei Balcani e del resto d’Europa ci ha permesso di descrivere lo scenario genetico della regione geografica dei Balcani.
Le misure della divergenza genetica tra coppie di popolazioni sono state rappresentate graficamente mediante Multidimensional Scaling. È stata inoltre effettuata un’Analisi delle Componenti Principali e un’analisi della varianza molecolare, AMOVA.
Lo studio è stato esteso anche a popolazioni del resto dell’Europa per comprendere se l’area dei Balcani rappresenti una realtà distinta dalle altre popolazioni europee oppure se la variabilità osservata concordi con quella europea.
Non sono state evidenziate significative differenze fra le popolazioni balcaniche e fra queste e altre popolazioni europee, anzi la variabilità genetica osservata e in accordo con quella osservata nel resto dell’Europa e pertanto, questo studio sembra suggerire che, nei Balcani, non ci sono correlazioni fra le differenze genetiche e le differenze linguistiche ed etniche.
Si può affermare che i Balcani, compresa la Serbia, e il resto dell’Europa, con poche eccezioni rappresentate da alcuni isolati come alcune popolazioni della costa e delle isole della Croazia, costituiscono un’entità genetica, a livello del mtDNA, piuttosto omogenea.Variabilità genetica in isolati linguistici: origine e struttura
Cristian Capelli (1), Francesca Scarnicci(2), Francesca Brisighelli(2,3), Federica Trombetta (2), Ilaria Boschi (2), Mara Masullo (2), Daniela Vantaggiato (2), Cinzia Grasso (2), Valerio Onofri (4), Donata Luiselli (5), Davide Pettener (5), Vincenzo L Pascali (2)
(1) Department of Zoology, University of Oxford, Oxford, UK (2)Istituto di Medicina Legale, Universita’ Cattolica del S. Cuore di Roma, Roma, Italia (3)Istituto di Medicina Legale, Universita’ di Santiago de Compostela, Santiago de Compostela, Spain (4) Istituto di Medicina Legale, Universita` Politecnica delle Marche, Ancona, Italia (5)Dipartimento di Biologia Evolutiva Sperimentale, Universita’ di Bologna, Bologna, Italia
Lo studio delle forze che influenzano la distribuzione della variabilità genetica fra popolazioni umane è un campo di ricerca di grande interesse per comprendere le dinamiche evolutive che determinano la struttura genetica delle popolazioni. La geografia appare come il fattore principale in questi processi, ma c’è una crescente mole di dati che suggerisce come anche la cultura possa avere un ruolo non trascurabile. Il linguaggio è senza dubbio uno degli aspetti culturali più caratterizzanti delle popolazioni umane. In passato è stata evidenziata una generale correlazione tra le maggiori famiglie linguistiche e le popolazioni umane, ma a livello continentale la geografia sembra essere l’elemento principale responsabile dell’attuale distribuzione di variabilità genetica fra popolazioni. È comunque da sottolineare che gli studi fino ad ora effettuati non si sono focalizzati in maniera specifica su questa problematica oppure non hanno tenuto in considerazione fattori confondenti come le distanze e le barriere geografiche, non rendendo possibile un’analisi appropriata dell’influenza delle lingue. In questo lavoro presentiamo lo studio della variabilità genetica di una serie di isolati linguistici e dei relativi vicini geografici, proponendo un approccio micro-geografico teso a minimizzare e controllare i fattori di disturbo e a focalizzare l’attenzione sul contributo delle lingue. Data la grande eterogeneità geografica, culturale e storica degli isolati linguistici presenti, l’Italia rappresenta un’area ottimale per questo tipo di studi. I dati genetici saranno discussi in funzione dell’origine degli isolati, dell’effetto delle lingue sul flusso genico e delle implicazioni a livello della struttura genetica.
Varianti del gene SLC24A5 in popolazioni umane di diversa origine geografica
E. Giardina, I. Pietrangeli, C. Martinez-Labarga, C. Martone, F. De Angelis, G.F. De Stefano, O. Rickards, G. Novelli
Dipartimento di Biologia, Università di Roma Tor Vergata Dipartimento di Biopatologia e Diagnostica per immagini, Università di Roma Tor Vergata
ABSTRACT MANCANTE